





















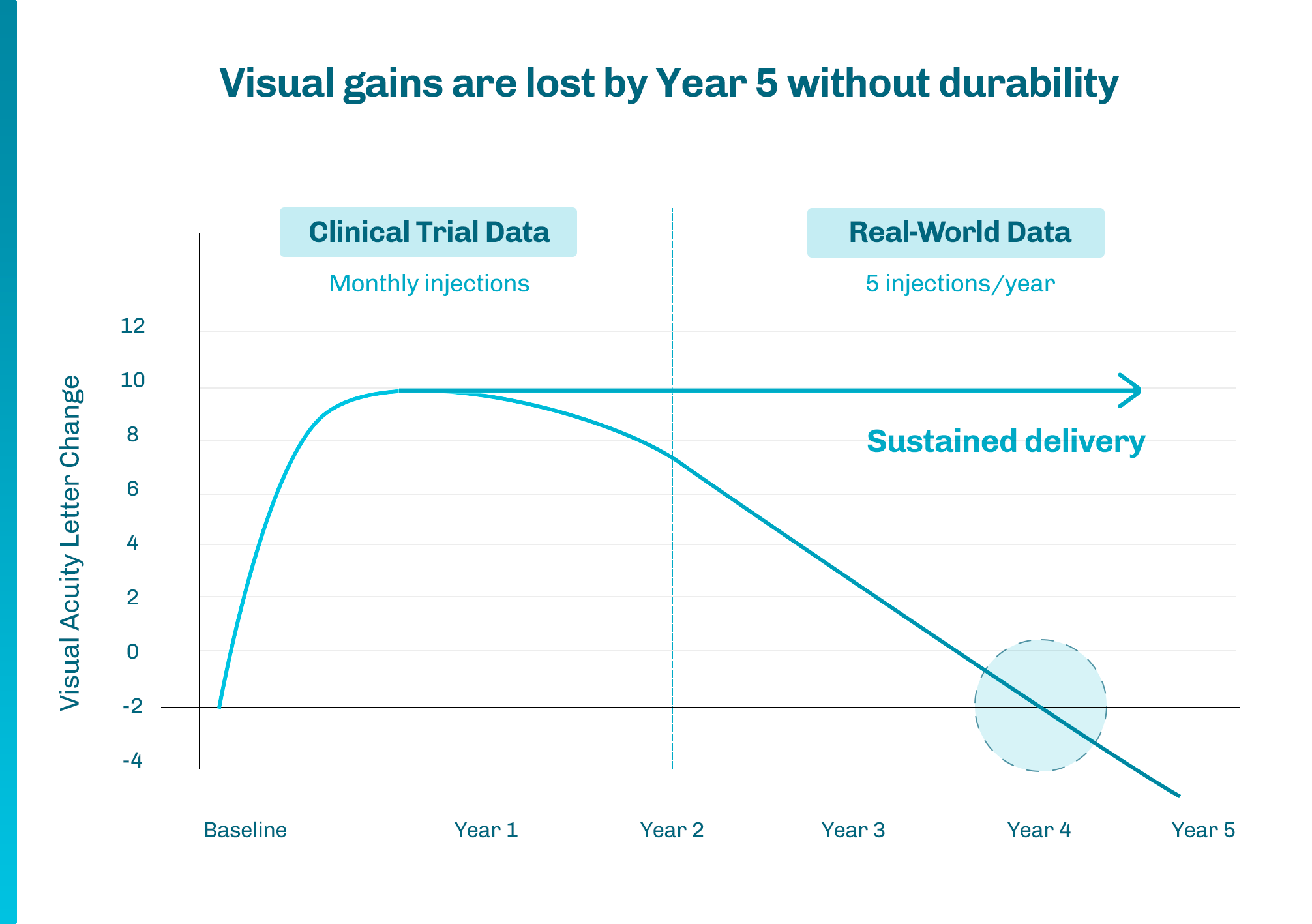
1 Khanani AM, et al. Ophthalmol Retina. 2020;4(2):122-133

JOB OPENINGS
We are a talented, passionate group of colleagues with a desire to translate innovative science into novel gene therapies for patients with neurodegenerative and ocular diseases and beyond.
We are committed to building a vibrant team combining deep expertise in AAV vector engineering and genetic construct design, innovative and advanced therapeutic product development, and manufacturing.
We are looking for more talented individuals to join our team.
Headquarters
63bis avenue Ledru Rolin
75012 Paris – France
Labs
Biolabs
Campus Broussais
Bâtiment Steg – 2e Etage
8 rue Helena Maria Vieira Da Silva
75014 PARIS
