






















![]()

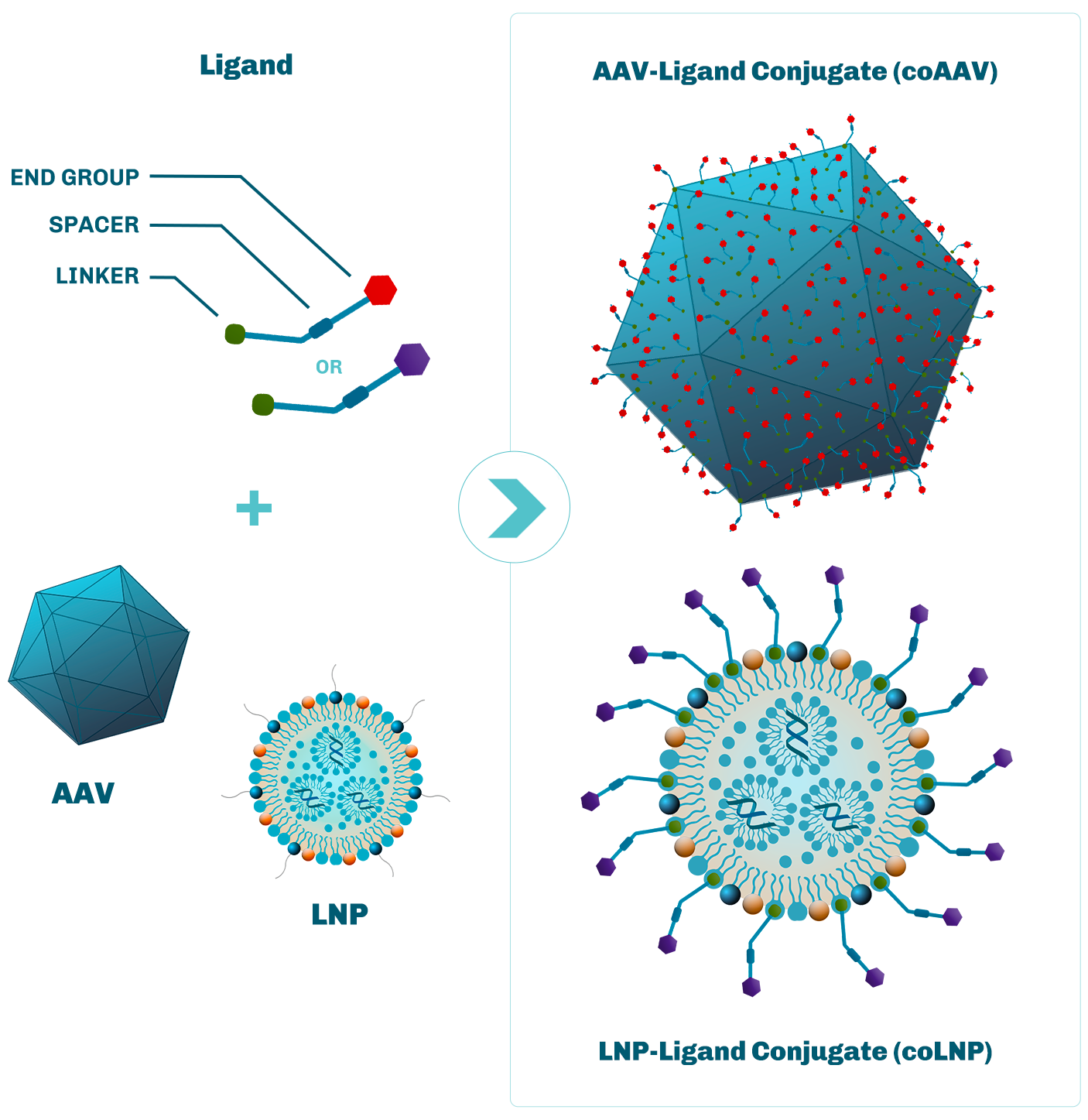

Site-specific conjugation onto capsid proteins enables altering of extracellular capsid sequestration, by blocking the binding of the AAV capsid to extracellular motifs.

Ligands are rationally designed to improve cell and tissue targeting, based on defined ligand-receptor interactions.

Site-specific conjugation of capsid proteins leads to improvement of intracellular AAV capsid trafficking and payload delivery to the nucleus.

Ligand conjugation reduces coAAV exposure to immune reaction and neutralizing antibodies.
Neurodegenerative disorders are a group of conditions characterized by progressive loss of structure or function of neurons, often leading to cognitive or motor impairment. These diseases, such as Alzheimer’s, Parkinson’s, and ALS, can arise from mutations that cause either a loss or gain of function in critical genes. Gene therapy offers a promising approach to target these underlying genetic causes. By delivering functional gene copies or silencing harmful ones, gene therapy can potentially restore normal cellular function.
Inherited retinal dystrophies are rare ophthalmic pathologies that can be divided into two groups:
Retinitis pigmentosa is the most common form of inherited retinal dystrophy representing 50% of all retinal dystrophies.
While multiple genes are implicated in each of these groups, within each patient or family, only one causative gene is involved.
May, 9 – 2025
April, 23 – 2025
April, 09 – 2025
January, 09 – 2025
December, 17 – 2024
October, 22 – 2024
September, 24 – 2024
September, 6 – 2024
May, 7 – 2024
April, 29 – 2024
April, 25 – 2024
March, 27 – 2024
February, 29 – 2024
October, 12 – 2023
September, 28 – 2023
September, 21 – 2023
May, 31 – 2023
May, 17 – 2023
May, 4 – 2023
March, 16 – 2023
October, 20 – 2022
September, 26 – 2022
September, 20 – 2022
September, 14 – 2022
September, 6 – 2022
June, 30 – 2022
June, 9 – 2022
May, 4 – 2022
April, 27 – 2022
April, 13 – 2022
November, 15 – 2021
October, 12 – 2021
September, 23 – 2021
September, 16 – 2021
July, 21 – 2021
JOB OPENINGS
We are a talented, passionate group of colleagues with a desire to translate innovative science into novel gene therapies for patients with neurodegenerative and ocular diseases and beyond.
We are committed to building a vibrant team combining deep expertise in AAV vector engineering and genetic construct design, innovative and advanced therapeutic product development, and manufacturing.
We are looking for more talented individuals to join our team.
Headquarters
63bis avenue Ledru Rolin
75012 Paris – France
Labs
INSTITUT DU CERVEAU ET DE LA MOELLE EPINIÈRE – ICM
Hôpital Pitié-Salpêtrière
47 bd de l’Hôpital
75013 Paris
